Trouxemos esta apresentação visto a importância de conhecer esse parâmetro na terapia por Ozônio do paciente.
OBJETIVIDADE DA APRESENTAÇÃO: G6PD X OZÔNIOTERAPIA
Todo e qualquer procedimento deve ser precedido de rigorosa anamnese, assim como ser precedido de análise técnica de procedimentos (riscos/benefícios).
A deficiência de glicose-6-fosfato desidrogenase (G6PD) é um distúrbio genético hereditário que pode resultar na destruição de glóbulos vermelhos (hemólise) depois de uma doença aguda ou uso de certos medicamentos.
A deficiência da G6PG – Glicose 6 Fostato Desidrogenase é a deficiência enzimática mais comum do mundo. Aproximadamente 400 milhões de pessoas são afetadas em todo mundo. Existe grande variabilidade de gravidade com base na mutação herdada.
BREVE RESUMO DA ENZIMA G6PD – G6PD – CLICOSE 6 FOSTATO DESIDROGENASE
A G6PD é uma enzima que contribui para o metabolismo da glicose com a consequente geração de energia.
É uma enzima citoplasmática presente em todas as células do corpo, que tem como função de catalisar a primeira etapa da rota metabólica da hexose monofosfato e, sendo assim, é essencial para a produção de NADPH, responsável por combater efeitos prejudiciais das espécies reativas de oxigênio e radicais livres
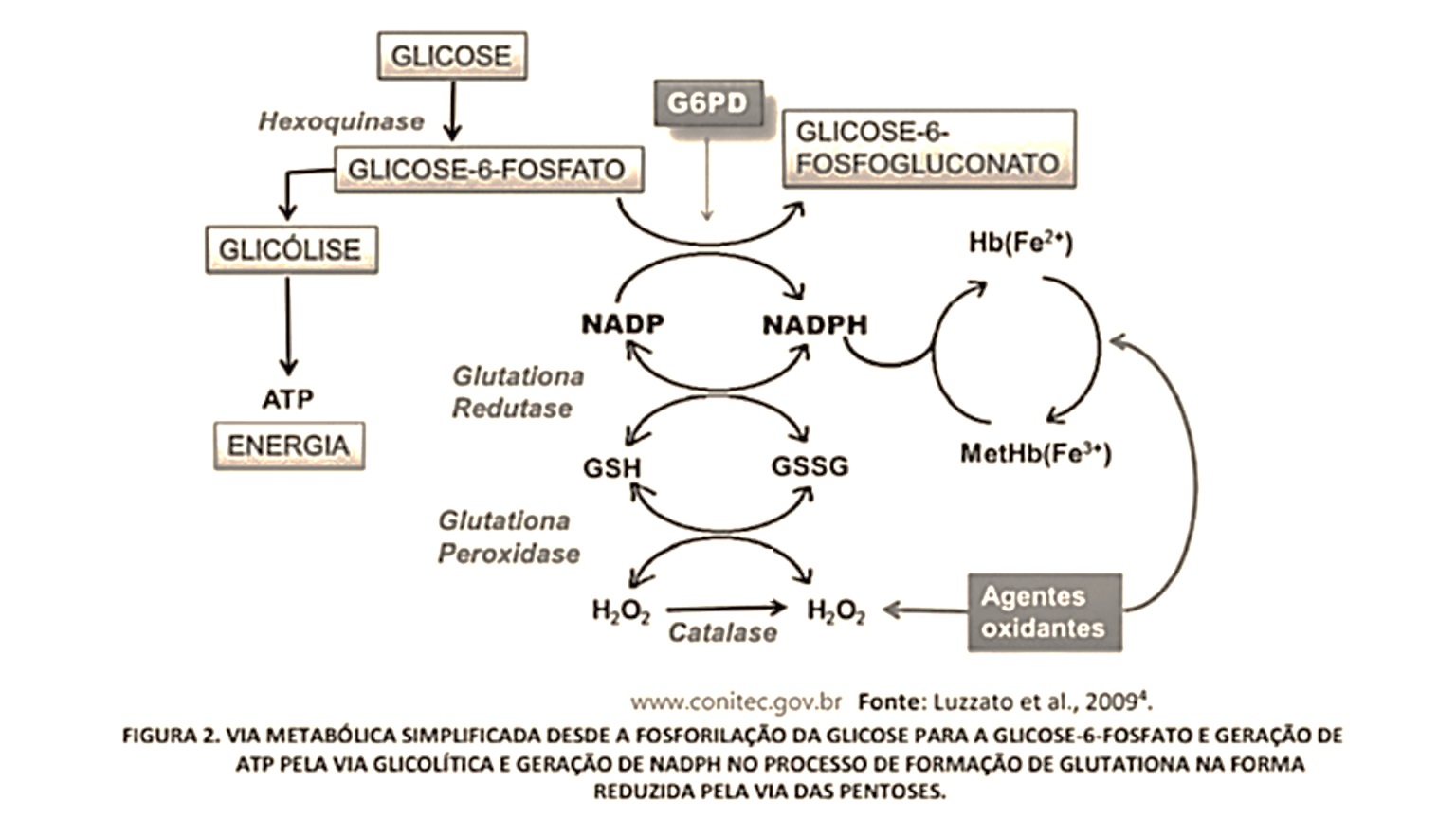
O eritrócito é uma célula particular porque transporta oxigênio para os tecidos sem consumi-lo. A adenosina trifosfato (ATP), energia calórica, e a nicotinamida adenina dinucleotídeo reduzida (NADH) e nicotinamida adenina dinucleotídeo fosfato reduzida (NADPH), energia redutora, são obtidas a partir da oxidação anaeróbica da glicose, via das pentoses e ciclo da glutationa.
A via das pentoses é responsável pela oxidação de cerca de 10% da glicose consumida pelo glóbulo vermelho. A ação de duas enzimas seqüenciais, a glicose-6-fosfato desidrogenase (G6PD) e a 6-fosfogliconato desidrogenase (6PGD) reduzem a coenzima nicotinamida adenina dinucleotídeo fosfato (NADP) à nicotinamida adenina dinucleotídeo fosfato reduzida (NADPH), sendo, praticamente, toda fonte geradora de NADPH. Ao manter a glutationa no estado reduzido (GSH), através da glutationa redutase, essa via desempenha importante papel na proteção do glóbulo vermelho frente aos processos oxidativos, que são responsáveis pela redução da vida média do eritrócito.
A deficiência de G6PD é a mais freqüente enzimopatia conhecida, afetando aproximadamente 400 milhões de pessoas no mundo. Os eritrócitos deficientes de G6PD são incapazes de reduzir NADP+ a NADPH em velocidade normal, apresentando assim um baixo potencial redutor, não conseguindo remover o peróxido de hidrogênio ou os dissulfetos mistos da hemoglobina formados após ingestão de certas drogas oxidantes e em alguns processos infecciosos e oxidativos. Ocorre hemólise autolimitada pela destruição dos eritrócitos senis, com conseqüente aumento de reticulócitos que apresentam maior quantidade da enzima.
A deficiência de G6PD é um modelo de interação genótipo-fenótipo, visto que os pacientes são, em sua maioria, assintomáticos, a não ser quando manifestaram o estresse oxidativo.
A grande maioria dos portadores são assintomáticos, e descobrem esta deficiência enzimática após o uso de determinados fármacos, alimentos ou corantes. A G6PD é uma enzima citoplasmática capaz de produzir substâncias que as protegem as células dos fatores oxidantes. A diminuição da atividade desta enzima afeta os eritrócitos tornando-os mais vulneráveis aos danos oxidativos, e a possibilidade de serem retirados da circulação em menos de 120 dias. Nesses momentos os pacientes apresentam anemia hemolítica aguda (AHA), uma manifestação hematológica importante.
Histórico
No século 19, pediatras gregos, italianos e portugueses observaram a ocorrência de anemia severa e hemoglobinúria em crianças após o consumo de feijões de fava, daí a alcunha de favismo. A observação mostrou que o favismo era recorrente nas mesmas pessoas e estava presente em famílias. Nos anos 1920, verificou-se que alguns pacientes, quando eram tratados para malária com primaquina e plasmoquina, apresentavam AHA. Não havia suspeita de que ambas as manifestações estavam conectadas.
Em 1956, um grupo de Chicago relatou que as hemácias das pessoas sensíveis à primaquina eram deficientes em G6PD. Pouco depois, em 1958, outro grupo em Genova na Itália encontrou a mesma deficiência em crianças com história anterior de favismo. Os achados combinados apreciaram que as duas manifestações foram causadas pela deficiência de G6PD e que essa era a genética e a herança foram transmitidas ao X.
Mecanismo de ação – Bases bioquímicas
A G6PD é uma enzima que ‘ORGANIZA SEU ORGANISMO” está presente em todas as células do organismo e age como a primeira enzima do ciclo das pentoses. Apesar de o produto final de sua reação ser a formação de uma pentose, é importante ressaltar que a sua atividade gera duas manifestações de NADPH, sendo esse seu trunfo principal. O NADPH é um doador de elétrons importantes em muitos processos celulares. Na maioria das células, outras enzimas também controlam NAPDH e, mesmo que haja deficiência de G6PD, isso não ocasionará problemas.
O mesmo não pode ser dito para as hemácias, que sacrificam muitas de suas organelas e enzimas na sua habilidade para exercer sua principal função de carregamento de oxigênio. Radicais livres podem ser produzidos no processo de transporte de oxigênio nas hemácias, nesse ponto o NADPH é fundamental para combater o estresse oxidativo.
A deficiência de G6PD nunca é completa e, caso fosse completa, seria fatal. A atividade residual da G6PD presente é capaz de manter as hemácias funcionando, mas não limita sua atividade. Quando ocorre um estresse oxidativo exógeno, as células não são capazes de aumentar sua produção de NADPH; consequentemente, a hemoglobina e outras proteínas são danificadas e as células são eventualmente abocanhadas por macrófagos ou sofrem hemólise devido ao dano.
Base genética da G6PD
O gene que codifica a enzima G6PD se encontra no braço longo dos cromossomos X (Xq28), o que demonstra a já mencionada herança ligada ao X. Isso acarreta algumas possibilidades fenotípicas.
A deficiência de G6PD não é mais comum em homens; apesar de mulheres homozigotas serem muito mais raras que homens hemizigotos, mulheres heterozigotas são muito mais numerosas. Por que isso é importante? Porque na prática clínica você vai encontrar mulheres heterozigotas onde o fenótipo de atividade da G6PD se apresenta como normal e em outras o fenótipo se mostra como a deficiência de G6PD, assim como visto nas homozigotas.
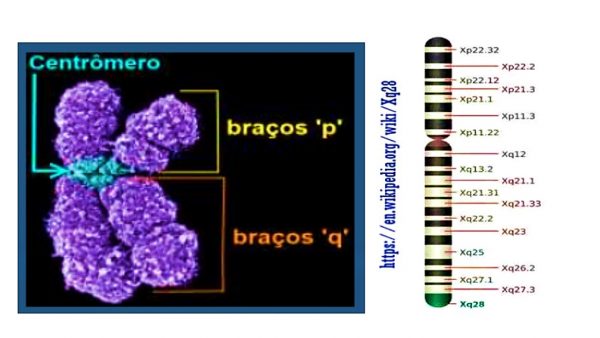
Os pares de cromossomos humanos são numerados de 1 a 22, sendo iguais para homens e mulheres.
É no 23º que está a diferença, ou seja, para os homens o par é X Y e para as mulheres é X X.
Cada cromossomo tem um braço curto chamado de “p” e um braço longo “q”. Além disso há várias subdivisões em bandas numeradas. A deficiência da G6PD é resultado de uma diferença no gene Xq28.
São mais de 300 variantes desta deficiência.
Manifestações clínicas
A deficiência de G6PD, apesar de na maior parte da vida das pessoas se manter sem repercussão, pode se manifestar clinicamente de três formas. É possível encontrar uma deficiência de G6PD como icterícia neonatal, anemia hemolítica aguda e anemia hemolítica crônica não esferocítica. A deficiência de G6PD torna o eritrócito suscetível ao estresse oxidativo, o que diminui a sobrevida dos eritrócitos. A hemólise ocorre depois de estresse oxidativo, comumente após febre, infecção bacteriana ou viral aguda e cetoacidose diabética. A hemólise é episódica e autolimitada, embora raros pacientes tenham hemólise crônica e contínua na ausência de estresse oxidativo.
Com menor frequência, a hemólise ocorre após a exposição a fármacos ou outras substâncias que produzam peróxido e causem oxidação da hemoglobina e membranas dos eritrócitos. A quantidade da hemólise depende do grau de deficiência de G6PD e do potencial oxidante do fármaco.
Exemplos:
Icterícia neonatal
Os recém-nascidos sempre apresentam um certo nível de hiperbilirrubnemia devido à necessidade de absorção da bilirrubina que antes era excretada pela placenta. Icterícia neonatal é definida como altos níveis de bilirrubinas para o peso e idade do recém-nascido. Se não for tratado, pode evoluir para o kernicterus, uma encefalopatia crônica que ocasiona atraso no desenvolvimento neuropsicomotor.
Uma das causas de icterícia neonatal é a deficiência de G6PD, mas não se sabe ao certo o seu mecanismo nessa ocorrência, e a deficiência de G6PD aumenta a incidência de icterícia em neonatos tanto do sexo feminino quanto masculino. Postula-se que neonatos deficientes de G6PD apresentam um turnover de heme maior, mas sem anemia ou com graus de anemia.
Anemia hemolítica aguda
A AHA é a apresentação mais clássica de deficiência de G6PD. Manifesta-se como episódios agudos de hemólise intravascular em um paciente previamente assintomático, geralmente após a ingestão de certos medicamentos ou alimentos e diante de infecção. Os sintomas da AHA são mal-estar, dor abdominal e lombar, hematúria, icterícia e palidez. Níveis elevados de LDH e manchas de sangue com anisocitose, policromasia e poiquilócitos são encontrados precocemente em episódios de AHA. Os reticulócitos também estão presentes como forma de resposta da medula óssea à hemólise. Favismo é o nome utilizado para AHA causado por feijão de fava, que contém divicina, um potente agente oxidante.
O curso da hemólise é normalmente autolimitado, possivelmente porque as células mais velhas (que apresentam níveis mais baixos de G6PD) são mais propensas à lise e são substituídas por células jovens que conseguem combater a agressão oxidativa.
Anemia hemolítica crônica não-esferocítica
A anemia hemolítica crônica não esferocítica é uma manifestação rara da deficiência de G6PD que ocorre em homens com variantes esporádicas da enzima. Ela se manifesta quando a hemácia não consegue manter níveis suficientes de NADPH para evitar o estresse oxidativo de seu próprio metabolismo. Por isso mantém um processo de hemólise crônica e o paciente apresenta icterícia, anemia, aumento de cálculos biliares, esplenomegalia e pode necessitar de suporte transfusional crônico. Os pacientes com a forma crônica podem apresentar quadros agudos quando expostos às mesmas que os outros pacientes, porém a dose necessária é geralmente menor para causar a crise hemolítica
Dosagem/ determinação da G6PD
1- Imagem
– Esfregaço periférico
A falha na função da G6PG diminui a geração de GLUTATIONA. Caso essas funções não ocorram, a hemoglobina da célula não irá se ligar ao O2, acabará por sofrer ruptura da membrana levando a hemólise. A desintegração do tetrâmero de hemoglobina gera polipeptídios desagregados precipitam-se no interior dos eritrócitos sob forma de corpos de Heinz.
– Teste de fluorescência
 O diagnóstico é feito sumariamente pela avaliação do teste de pontos de fluorescência ou análise quantitativa da atividade de G6PD por espectrofotometria. O teste de fluorescência é rápido e barato, porém ele pode apresentar falso negativo em mulheres heterozigotas, visto que as hemácias são as que não acompanham hemólise e, por sua característica de mosaico, esses pacientes apresentam um teste normal. Alguns pacientes e famílias se beneficiam da avaliação genética da mutação para descrição, mas esses testes são sumariamente utilizados em pesquisa.
O diagnóstico é feito sumariamente pela avaliação do teste de pontos de fluorescência ou análise quantitativa da atividade de G6PD por espectrofotometria. O teste de fluorescência é rápido e barato, porém ele pode apresentar falso negativo em mulheres heterozigotas, visto que as hemácias são as que não acompanham hemólise e, por sua característica de mosaico, esses pacientes apresentam um teste normal. Alguns pacientes e famílias se beneficiam da avaliação genética da mutação para descrição, mas esses testes são sumariamente utilizados em pesquisa.
Ex: Deficiência hereditária de eritrócitos G6PD
O esfregaço de sangue é de um homem de 37 anos com deficiência hereditária de G6PD. Mostra muitas células bolhosas (flechas vermelhas) devido à precipitação da hemoglobina lesada no interior das células. Também mostra esferócitos (flechas azuis) com danos à membrana decorrente da ligação pela hemoglobina oxidada.
Imagem cedida por Jerry L. Spivak, MD.
2- Dosagem da G6PD
A dosagem laboratorial da G6PD é uma prática bastante simples. Coleta em franco com EDTA
CBHPM 4.03.02.05-9 AMB 28.01.098-
Preparo do Paciente: Jejum não obrigatório.
Método: Determinação da ação do G6PD em lisado de hemácias, em soro ou em plasma por sua ação sobre a glicose 6 fosfato.
Interpretação:
DIMINUIÇÃO: deficiência de G6PD (hemólise secundária, congênita, desencadeada por drogas ou por infecções virais e bacterianas).
AUMENTO e NORMAL: normalmente não tem significado clínico.
- O método utilizado para a determinação quantitativa da atividade da G-6-PD utilizado no laboratório é baseado na oxidação da glicose-6- fosfato (G-6-P) a 6-fosfogliconato (6-PGA) e na redução do NADP a NADPH, analisado por espectrofotometria no comprimento de onda a 340nm, conforme protocolo publicado por Beutler em 1984.
Valor de Referência: de 6,97 a 20,50 U/g hemoglobina
Fonte: Deficiência de glicose-6-fosfato desidrogenase na icterícia neonatal: avaliação de 10 anos do Instituto Adolfo Lutz-SP // Marilena OSHIRO; Vânia Maria CAÇÃO; Cristiani Martinez SALZONE Núcleo de Hematologia e Bioquímica -Centro de Patologia-Instituto Adolfo Lutz // Bol Inst Adolfo Lutz. 2014; 24(1):33-35 • 33
Sinais e sintomas:
FAVISMO – DEFICIÊNCIA DE G6PD – DOENÇA – https://www.msdmanuals.com/
Quando os sintomas se desenvolvem, incluem:
§ febre § urina escura. § dor abdominal. § dor nas costas. § fadiga. § palidez na pele. A maioria das pessoas se recupera em poucos dias sem tratamento em acometimento agudo.
Controle da G6PD
O gerenciamento da deficiência de G6DP é sobretudo preventivo.
Após o diagnóstico, é muito importante orientar o paciente sobre a lista de medicamentos e alimentos que não podem ser consumidos por esses pacientes. Condutas de educação são fundamentais.
Cada uma das apresentações da doença tem seu manejo específico. A icterícia neonatal é tratada da mesma forma que outros tipos de icterícia, através de fototerapia ou exsanguinotransfusão em casos mais graves. A AHA é inicialmente tratada através do tratamento do fator causador do estresse oxidativo, seja a suspensão de medicamentos ou ingestão de alimentos ou o tratamento infeccioso apropriado. Em casos em que a anemia se torna grave, pode-se utilizar de transfusão de concentrado de hemácias. Para a forma crônica da doença, o tratamento preventivo é ainda mais importante, visto que esses pacientes podem sofrer complicações graves em quadros de agudização.
OBJETIVIDADE DA APRESENTAÇÃO: G6PD X OZONIOTERAPIA
POR QUE É IMPORTNATE DOSAR A G6PD ANTES DO PROCEDIMENTO DE OZONIO?
Como explicado, a deficiência de glicose-6-fosfato desidrogenase (G6PD) é a doença hereditária mais comum do metabolismo dos eritrócitos e pode causar hemólise na presença de gatilhos. A incidência é mais alta em certos grupos étnicos (p. ex., afro-americanos, pessoas de origem mediterrânea, pessoas de descendência asiática). Os gatilhos incluem doenças agudas (p. ex., infecções), fármacos (p. ex., salicilatos) e outras substâncias (p. ex., favas) que causam estresse oxidativo.
A deficiência de G6PD é uma doença que muito bem demonstra a interação genótipo-fenótipo e o conceito de farmacogenética, pela sua manifestação clínica a partir da exposição ao estresse oxidativo.
O corpo humano, mediante a ativação de leucócitos, pode produzir ozônio – grande aliado da saúde em situações normais e patológicas. Ele reage com biomoléculas – poli-insaturados (PUFA) e antioxidantes – presentes no plasma (um dos componentes do sangue).
AO MINISTRARMOS OZÕNIO:
A reação entre peróxido de hidrogênio e outras possibilidades, os ROS (espécies reativas de oxigênio) e produtos de peroxidação lipídica (LOPs), levam a um súbito aumento da concentração de peróxido de hidrogênio, gerando um gradiente que acarreta sua rápida transferência para dentro das células. Depois, em poucos segundos, serão ativados vários processos bioquímicos que, simultaneamente, sofrerão redução para água pelo eficiente sistema antioxidante intracelular (GSH, catalase e GSH-Px).
Essa etapa crítica corresponde a um estresse oxidativo controlado, agudo e transitório necessário para ativação biológica – sem toxicidade concomitante, provando que a dosagem de ozônio é compatível com a capacidade antioxidante do sangue.
Enquanto as ROS são responsáveis pelos efeitos biológicos imediatos, os LOPs são importantes por seu efeito tardio, quando o sangue ozonizado retorna para a circulação por reinfusão. Os LOPs podem chegar a qualquer órgão após se ligarem ao receptor suscitando a adaptação para repetidos estresses oxidativos agudos. Eles ativarão a regulação das enzimas antioxidantes e, provavelmente, a liberação de células-tronco.
Esse controle oxidativo sem a presença da G6PD, em um ambiente onde, pela falta da G6PD já está altamente oxidado, gerará uma cascata que, se não controlada, poderá trazer danos irreparáveis.
Resumo: Com o aumento do estresse oxidativo, pode diminuir a geração de GLUTATIONA. Caso essas funções não ocorram, a hemoglobina da célula não irá se ligar ao O2, acabará por sofrer ruptura da membrana levando a hemólise.
G6PD BAIXA : significa que o indivíduo está em estresse oxidativo!
G6PD abaixo de 2,2 é aconselhável não oxidar mais o organismo, (ozônio é altamente oxidante), devemos tratar o estresse com anti oxidantes, vitamina C, vitamina E, coenzima Q10, Glutationa, etc.
Complemento:
Lista de medicamentos e alimentos não recomendados para pacientes com deficiência de G6PD.
Analgésicos:
Aspirina, acetofenitidina, probenecida
Antimaláricos
Cloroquina, hidroxicloroquina, primaquina, quinino
Citotóxico/Antibacterianos
Cloranfenicol, sulfametoxazol+trimetropim, ácido nalidíxico, nitrofurantoína
Drogas Cardiovasculares
Dapsona, sulfametoxipirimidina, sulfapiridina, sulfanilamida, sulfasalazina
Outros compostos
Alfa-metildopa, ácido ascórbico, hidralazina, azul de metileno, naftaleno (naftalina), urato oxidase, Vitamina K
Alimentos
Feijão de fava, água tônica, vinho tinto, mirtilos (mirtilo)
Célia Wada – CRF-SP – 7043
Referências:
- Luzzatto L, Nannelli C, Notaro R. Deficiência de glicose-6-fosfato desidrogenase. Vol. 30, Clínicas de Hematologia/Oncologia da América do Norte. WB Saunders; 2016. pág. 373–93.
- Cappellini MD, Fiorelli G. Seminário Deficiência de glicose-6-fosfato desidrogenase [Internet]. Vol. 371, www.thelancet.com. 2008. Disponível em: www.thelancet.com
- Associazione Italiana Favismo – www.g6pd.org
- Mason PJ, Bautista JM, Gilsanz F. Deficiência de G6PD: a associação genótipo-fenótipo. Análises de Sangue. 2007;21(5):267–83.
- Luzzatto L, Seneca E. Deficiência de G6PD: Um exemplo clássico de farmacogenética com implicações clínicas contínuas. Vol. 164, British Journal of Hematology. 2014. pág. 469–80.
- Atividade da 6-fosfogliconato desidrogenase em deficientes de glicose Activity of 6-phosphogluconate dehydrogenase in glucose-6-phosphate dehydrogenase deficiency – Daniela B. NicolieloRosecler I.P. FerreiraAmauri A. Leite https://www.scielo.br/j/rbhh/a/JncptYxwT763Pps5GqkMzfP/?lang=pt#:~:text=As%20enzimas%20G6PD%20e%206PGD,estresse%20oxidativo%20metab%C3%B3lico%20nos%20eritr%C3%B3citos.
- Deficiência de glicose-6-fosfato desidrogenase na icterícia neonatal: avaliação de 10 anos do Instituto Adolfo Lutz-SP – Marilena OSHIRO; Vânia Maria CAÇÃO; Cristiani Martinez SALZONE Núcleo de Hematologia e Bioquímica -Centro de Patologia-Instituto Adolfo Lutz – Bol Inst Adolfo Lutz. 2014; 24(1):33-35 • 33
- Leite AA. Icterícia neonatal e deficiência de glicose-6-fosfato desidrogenase. Rev Bras Hematol Hemoter. 2010; 32(6):430- 431.
- Algur N, Avraham I, Hammerman C, Kaplan M. Quantitative Neonatal Glucose-6-Phosphate Dehydrogenase Screening: Distribution, Reference Values, and Classification by Phenotype. J Pediatric. 2012; 161(2):197-200.
- Kaplan M, Hammerman C. Glucose-6-phosphate dehydrogenase deficiency and severe neonatal hyperbilirubinemia: a complexity of interactions between genes and environment. Semin Fetal Neonatal Med. 2010; 15:148-156.
- WHO Working Group. Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ. 1989; 67(6):601-611.
- Laboratório Hérmes Pardini.
